g*****n
发帖数: 250 | 1 韩春雨的论文这么火,我这个外行也忍不住要拜读一下。外行就看图识字,直接奔图版
。可没想到,第一张图(Fig. 1a) 就给我一棍。图标说“(a) Electrophoresis of
GST-NgAgo expressed in E. coli, purified by Sephrose-4B, digested by protein
kinase K, and treated with the indicated exonucleases.” 这显然是用激酶磷酸
化的蛋白了(虽然“digested"这个词怪点)。可图上是“Single Strand (DNA)
molecular weight marker”。??? 只好到文字里看看。啊,原来是作者粗心,审稿
粗心,编辑粗心。
第一张图算是连猜带蒙过去了。看第二张图 (Fig. 1b) 吧,更糊涂了。一个quide在,
只切质粒的一条链 (OC)。只有两个quide同时在时, 才切质粒的两条链 (LIN)。可作者
在摘要里声称一个quide就能切质粒的两条链啊。看来我不仅是外行,而且又笨。算了
,不往下看了。 |
F****n
发帖数: 5117 | 2 看不懂不要紧,倒数第二句的结论对了就好
protein
,
【在 g*****n 的大作中提到】
: 韩春雨的论文这么火,我这个外行也忍不住要拜读一下。外行就看图识字,直接奔图版
: 。可没想到,第一张图(Fig. 1a) 就给我一棍。图标说“(a) Electrophoresis of
: GST-NgAgo expressed in E. coli, purified by Sephrose-4B, digested by protein
: kinase K, and treated with the indicated exonucleases.” 这显然是用激酶磷酸
: 化的蛋白了(虽然“digested"这个词怪点)。可图上是“Single Strand (DNA)
: molecular weight marker”。??? 只好到文字里看看。啊,原来是作者粗心,审稿
: 粗心,编辑粗心。
: 第一张图算是连猜带蒙过去了。看第二张图 (Fig. 1b) 吧,更糊涂了。一个quide在,
: 只切质粒的一条链 (OC)。只有两个quide同时在时, 才切质粒的两条链 (LIN)。可作者
: 在摘要里声称一个quide就能切质粒的两条链啊。看来我不仅是外行,而且又笨。算了
|
g*****n
发帖数: 250 | 3 忍不住, 还是要继续读。
Fig.1c跟Fig.1b相互矛盾。这两个实验条件是一样的,可是,1c, 一个guide切两条链
。而1b, 一个guide切一条链。这两个不能都对,但有可能都错。
Fig.3a, NgAgo在细胞里把质粒100%切开,胶上是线型的(LIN),没有被修复。可是,却
能测序 (Fig.3b)。 |
C****5
发帖数: 32 | |
g*****n
发帖数: 250 | 5 嗯,对。是TA克隆。他写在前面了。
就是说,切开了,不修复。这样的话,就不是editing了,是cutting/damaging。那Fig
.3d就不是editing的结果。 |
g*****n
发帖数: 250 | 6 继续读。
Figure 5。用NgAgo切掉两个荧光蛋白基因之间的STOP codon,使两个蛋白成为融合蛋
白。这个必须在stop这个位置切掉三个核苷酸,使两个编码区in-frame。多一个不行,
少一个也不行。
Figure 3显示,NgAgo随机地切掉数量不等的核苷酸。在24nt guided区域里,所切得的
产物类型应该是很多,很多,很多。可以想象,在这样24nt区域里,既要准确地在stop
位置上,又要只切掉三个。懂数学的给算算,这个机率将是多少。(这还没把NHEJ带来
的错误算上)。感觉有点天方夜谭。作者好像心里也没底。“Imprecise genome
repair mediated by nonhomologous end joining (NHEJ) mutated the TGA
terminator and made the expression of mRFP-eGFP chimera possible” 这么关键
的实验,没有测序确定,就一句猜测性话。 |
l*****7
发帖数: 8463 | 7 什么时候能读完论文?
想拜读你的读书报告或读后感。 |
g*****n
发帖数: 250 | 8 接着看Figure 5. 虽说准确地切掉stop犹如登天,作者还是要撞撞运气,跳过中间步骤
,直接测荧光。如果测到绿荧光,就表示eGFP in frame, 并表达。作者用flow
cytometry测量有绿荧光的细胞。具体怎么做的,不得而知,因为作者没有描述。从结
果上看,应该是,分别用RFP和GFP的激发波长来激发红(纵坐标)和绿(横坐标)荧光
。到这儿,问题出来了。RFP和GFP融合成一体后,两个蛋白会互相传递能量,即FRET效
应。这时,融合蛋白的最佳激发波长既不是RFP的,也不是GFP的波长。用GFP的波长来
测,显然脱靶了。 |
C****5
发帖数: 32 | 9 我也仔细的去看了一下FIGURE5A,文章中敲入一个几K的序列进去,两端的同源臂只有15
个nt。即使NGAGo能够100%的切开基因组,这KI的效率也几乎为0吧。根本就不会去这样
设计。
做过KI的大概都对要多长的同源臂有一个认识。
还有FIGURE5c, 正如楼上说的, 文章中要精确的切掉TAG这三个碱基, HAN设计的
gDNA 居然在这三个碱基前面。
按照正常的逻辑,肯定是会选择包含着三个碱基的gDNA.
理论上,这么低的概率的事情, 根本不会用来做ASSAY. 但是惊人的,效率高达11.7% |
C****5
发帖数: 32 | 10 而且 HAN还应该吧那11.7%的细胞拿去测序。也没有。 |
|
|
g*****n
发帖数: 250 | 11 那11.7%是什么,现在也成了谜了。If something sounds too good to be true, it
must be too good to be true. 那11.7%怎么来的,只有作者知道。现在流行一句话:
“挤一挤,总会有的“。 |
g*****n
发帖数: 250 | 12 回来看Figure 2。NgAgo的表达质粒和guide一起转染到细胞里,48小时后提纯NgAgo蛋
白,然后走胶测guide (Figure 2a)。转染100-300ng guide,转染率不会是100%,回
收率也不会是100%,经过这么多的步骤还能看到guide?
更惊人的是,先转染质粒,12-24小时后再转染guide (Figure 2b)。这些guide 有多少
能恰好进到含有质粒的细胞里。经过这么多的步骤还能看到guide? |
c********6
发帖数: 693 | 13 编的文章就是这样,有的时候逻辑会出问题
【在 g*****n 的大作中提到】
: 回来看Figure 2。NgAgo的表达质粒和guide一起转染到细胞里,48小时后提纯NgAgo蛋
: 白,然后走胶测guide (Figure 2a)。转染100-300ng guide,转染率不会是100%,回
: 收率也不会是100%,经过这么多的步骤还能看到guide?
: 更惊人的是,先转染质粒,12-24小时后再转染guide (Figure 2b)。这些guide 有多少
: 能恰好进到含有质粒的细胞里。经过这么多的步骤还能看到guide?
|
c********6
发帖数: 693 | 14 编的文章就是这样,有的时候逻辑会出问题
【在 g*****n 的大作中提到】
: 回来看Figure 2。NgAgo的表达质粒和guide一起转染到细胞里,48小时后提纯NgAgo蛋
: 白,然后走胶测guide (Figure 2a)。转染100-300ng guide,转染率不会是100%,回
: 收率也不会是100%,经过这么多的步骤还能看到guide?
: 更惊人的是,先转染质粒,12-24小时后再转染guide (Figure 2b)。这些guide 有多少
: 能恰好进到含有质粒的细胞里。经过这么多的步骤还能看到guide?
|
P****R
发帖数: 22479 | 15 警察审讯嫌疑犯,首先要他描述事情经过,然后让他倒过来描述事情经过。
编假的嫌疑犯往往无法重复描述准确。
【在 c********6 的大作中提到】
: 编的文章就是这样,有的时候逻辑会出问题
|
s*********y
发帖数: 292 | 16 LZ看的很仔细,我感觉他造都不太会造。
像这种正好切掉3个nt的STOP codon的实验,实验设计的时候就不应该决定去做
GFP-RFP fusion之后FRET,GFP的能量都漏给RFP去了,不过一般GFP channel也能测到
,不会100%消失的。但还是那句话,这实验压根就不应该这么设计
总之就是水平有限,没发过大文章也搞不清楚大文章发出来之后的后果。他发文章之前
可能想着把文章搞出来评评职称、交交差,压根就没有想到发出来之后会被那么多媒体
吹到天上去,然后被那么多重复不出来的人揪着不放。现在是骑虎难下了 |
g*****n
发帖数: 250 | 17 对于Figure 4, 大家都在等测序的结果,我就没细看。看到对4a的讨论,我就细看看。
对4a质疑是有道理的。G5和G13相差几十个nt,胶上应该看得出差别。
另外,按Figure 3,如果NgAgo随机切,在一个guided 24nt区域里应该可以切到相差十
几个nt的产物。在PAGE在样高分辨的胶上是应该看得到。即使不会是清晰的带,多带倾
向现象应该看得出来。可是,作者所有测试的基因都是两条产物带。
Figure 4d, 两个黑箭头指给读者看两条产物带。可是,在这两条带之间还有一条带(
300左右)。这条带和250下边的产物带一样厚,作者确不理了。这条带很难用污染或非
特异来解释,因为,那些PCR产物都是纯化后才做T7E1的,而且每个样品都有。
有些不解的是,Supplementary Figure 10显示,作者用一对引物可得到两条清晰的带
。很好的引物。可是,在正文的实验里,作者却用了另外一对引物,而得到一条模糊的
带和“非特异”带。 |
z*******4
发帖数: 285 | 18 从你提出的质疑来看,你太外行,以你现有的知识无法与你讨论。
作为外行,请你尊重一下生物科研,请尊重一下Nature Biot杂志的编辑,以及审稿的
行业权威。如果你一个外行都能看出这么多问题而文章顺利发表,这些编辑审稿同行都
可以去死了。
你不懂不是你的错,你不懂还要出来说别人错了,那就是你的问题了
protein
,
【在 g*****n 的大作中提到】
: 韩春雨的论文这么火,我这个外行也忍不住要拜读一下。外行就看图识字,直接奔图版
: 。可没想到,第一张图(Fig. 1a) 就给我一棍。图标说“(a) Electrophoresis of
: GST-NgAgo expressed in E. coli, purified by Sephrose-4B, digested by protein
: kinase K, and treated with the indicated exonucleases.” 这显然是用激酶磷酸
: 化的蛋白了(虽然“digested"这个词怪点)。可图上是“Single Strand (DNA)
: molecular weight marker”。??? 只好到文字里看看。啊,原来是作者粗心,审稿
: 粗心,编辑粗心。
: 第一张图算是连猜带蒙过去了。看第二张图 (Fig. 1b) 吧,更糊涂了。一个quide在,
: 只切质粒的一条链 (OC)。只有两个quide同时在时, 才切质粒的两条链 (LIN)。可作者
: 在摘要里声称一个quide就能切质粒的两条链啊。看来我不仅是外行,而且又笨。算了
|
L*****t
发帖数: 56 | 19 呵呵,小保方和韩的大作都是反复投science未果而投nature(子刊)一发命中,整个过
程一致到不像巧合的程度。
韩的事情先不加评论,小保方投science的审稿人意见后来被人贴到了网上[0],里面基
本上把对这篇文章语焉不详和自相矛盾之处都指出来了,事实证明后来对STAP细胞的怀
疑依然是围绕这些问题点展开最后证明其造假。后来这篇文章为什么会发在Nature上谁
也说不清楚,但是与其相信审稿人集体智商断线,编辑被猪油糊了心无视批评抢发稿的
可能性明显更大。至于韩自称审稿人自行重复部分实验结果这个就更离奇了.
[0] http://retractionwatch.com/2014/09/10/truly-extraordinary-simply-not-credible-suspiciously-sharp-a-stap-stem-cell-peer-review-report-revealed/
【在 z*******4 的大作中提到】
: 从你提出的质疑来看,你太外行,以你现有的知识无法与你讨论。
: 作为外行,请你尊重一下生物科研,请尊重一下Nature Biot杂志的编辑,以及审稿的
: 行业权威。如果你一个外行都能看出这么多问题而文章顺利发表,这些编辑审稿同行都
: 可以去死了。
: 你不懂不是你的错,你不懂还要出来说别人错了,那就是你的问题了
:
: protein
: ,
|
g*****n
发帖数: 250 | 20 我没有请你来讨论,别抬高自己。
我对编辑和审稿没有恩怨,不必攻击他们,也不必尊重他们。
在科学上没有权威。
【在 z*******4 的大作中提到】
: 从你提出的质疑来看,你太外行,以你现有的知识无法与你讨论。
: 作为外行,请你尊重一下生物科研,请尊重一下Nature Biot杂志的编辑,以及审稿的
: 行业权威。如果你一个外行都能看出这么多问题而文章顺利发表,这些编辑审稿同行都
: 可以去死了。
: 你不懂不是你的错,你不懂还要出来说别人错了,那就是你的问题了
:
: protein
: ,
|
|
|
P****R
发帖数: 22479 | 21
就事论事,假定韩的结果是真实的,应该可以重复。如果无法重复,N+1都证明无
法重复。韩就应该实事求是,撤稿,继续埋头苦干10年。
【在 g*****n 的大作中提到】
: 我没有请你来讨论,别抬高自己。
: 我对编辑和审稿没有恩怨,不必攻击他们,也不必尊重他们。
: 在科学上没有权威。
|
m****a
发帖数: 270 | 22 为啥要刚好切掉3个nt?文章中的target site 在stop前面(Figure 5c,
Supplementary Figure 7)
切割后indel导致 reading frame shift,会有1/3的概率表达GFP
EGFP-linker-mRFP 的FRET效率低于0.1,mRFP的存在对GFP荧光影响不大。
而且同时给两种激发光的情况下,不需要考虑FRET效应。
从理论上说如果11%的细胞表达GFP,那么实际的编辑效率可能高达33%。
就想CK2015所说,这么高的效率插入将近2.5kb的片段,光靠两边15bp的同源臂简直天
方夜谭。除非阿狗会变魔术。
【在 s*********y 的大作中提到】
: LZ看的很仔细,我感觉他造都不太会造。
: 像这种正好切掉3个nt的STOP codon的实验,实验设计的时候就不应该决定去做
: GFP-RFP fusion之后FRET,GFP的能量都漏给RFP去了,不过一般GFP channel也能测到
: ,不会100%消失的。但还是那句话,这实验压根就不应该这么设计
: 总之就是水平有限,没发过大文章也搞不清楚大文章发出来之后的后果。他发文章之前
: 可能想着把文章搞出来评评职称、交交差,压根就没有想到发出来之后会被那么多媒体
: 吹到天上去,然后被那么多重复不出来的人揪着不放。现在是骑虎难下了
|
m****a
发帖数: 270 | 23 仔细看了一下,文章中并没有给出具体knockin效率是多少, 作者用了G418筛选,
所以使用15bp的同源臂即使只有万分之一的重组概率也是有可能筛到stable cell line。
不用长一点同源臂不合常理,为什么这么搞,得去问韩春雨。
15
【在 C****5 的大作中提到】
: 我也仔细的去看了一下FIGURE5A,文章中敲入一个几K的序列进去,两端的同源臂只有15
: 个nt。即使NGAGo能够100%的切开基因组,这KI的效率也几乎为0吧。根本就不会去这样
: 设计。
: 做过KI的大概都对要多长的同源臂有一个认识。
: 还有FIGURE5c, 正如楼上说的, 文章中要精确的切掉TAG这三个碱基, HAN设计的
: gDNA 居然在这三个碱基前面。
: 按照正常的逻辑,肯定是会选择包含着三个碱基的gDNA.
: 理论上,这么低的概率的事情, 根本不会用来做ASSAY. 但是惊人的,效率高达11.7%
|
g*****n
发帖数: 250 | 24 如果切掉的不是3个,后面的编码就变了,就不是eGFP了。那个stop codon (正文里是
TGA,Supplementary Figure 7里是TAG)不在靶区,这个设计的问题就更大了。要想破
坏它,就只能移位,后面的编码就必定乱。无论如何,用这个方法来破坏stop,又要使
eGFP in frame就是天方夜谭。
你怎么知道“EGFP-linker-mRFP 的FRET效率低于0.1”?这个取决于linker的大小和性
质。作者并没有讨论这个。无论如何,这个形象存在,实验上就不可不考虑。
【在 m****a 的大作中提到】
: 为啥要刚好切掉3个nt?文章中的target site 在stop前面(Figure 5c,
: Supplementary Figure 7)
: 切割后indel导致 reading frame shift,会有1/3的概率表达GFP
: EGFP-linker-mRFP 的FRET效率低于0.1,mRFP的存在对GFP荧光影响不大。
: 而且同时给两种激发光的情况下,不需要考虑FRET效应。
: 从理论上说如果11%的细胞表达GFP,那么实际的编辑效率可能高达33%。
: 就想CK2015所说,这么高的效率插入将近2.5kb的片段,光靠两边15bp的同源臂简直天
: 方夜谭。除非阿狗会变魔术。
|
m****a
发帖数: 270 | 25 你能注意到正文里的STOP codon和supplemental 里的不一样说明你眼神不错。
但是RFP和GFP之间相差了100bp,你仔细再多数几遍。100=33X3+1, 正确的移码突变应
该在stop前切除 1, 4, 7, 10.....个bp。
EGFP和mRFP之间FRET效率多少你自己去查文献,关键不在这里,Acceptor 通道直接用
激光激发了,还需要用 Donor 提供能量么? 况且这个方法又不是韩春雨发明的,
surrogate reporters早在TALEN、ZFN时代就已经有人用了。
http://nature.com/nmeth/journal/v8/n11/full/nmeth.1733.html
https://tools.thermofisher.com/content/sfs/posters/Use-of-surrogate-reporter
-vectors-to-enrich-for-CRISPR-and-TALEN-modified-cells.pdf
http://pnabio.com/products/Reporter.htm
【在 g*****n 的大作中提到】
: 如果切掉的不是3个,后面的编码就变了,就不是eGFP了。那个stop codon (正文里是
: TGA,Supplementary Figure 7里是TAG)不在靶区,这个设计的问题就更大了。要想破
: 坏它,就只能移位,后面的编码就必定乱。无论如何,用这个方法来破坏stop,又要使
: eGFP in frame就是天方夜谭。
: 你怎么知道“EGFP-linker-mRFP 的FRET效率低于0.1”?这个取决于linker的大小和性
: 质。作者并没有讨论这个。无论如何,这个形象存在,实验上就不可不考虑。
|
P****R
发帖数: 22479 | 26 韩现在做缩头乌龟。
line。
【在 m****a 的大作中提到】
: 仔细看了一下,文章中并没有给出具体knockin效率是多少, 作者用了G418筛选,
: 所以使用15bp的同源臂即使只有万分之一的重组概率也是有可能筛到stable cell line。
: 不用长一点同源臂不合常理,为什么这么搞,得去问韩春雨。
:
: 15
|
P****R
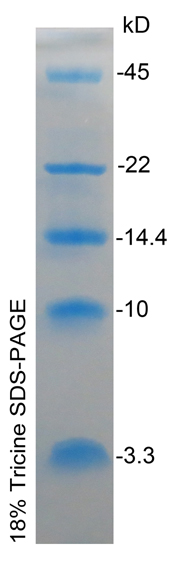
发帖数: 22479 | 27 关于电泳条带的拖尾问题
方先生:
您好!
我是您的超级粉丝,从“基因皇后”事件就开始关注新语丝,一直到现在,
只要打开浏览器,肯定要到新语丝上看一看。
看了7月20号关于韩春雨的文章,不谈其它,我只就电泳条带的拖尾现象谈
一下我的认识。
一般情况下,电泳条带拖尾状况正如方先生所说,边缘拖后,中间超前。我
原先也是一直是这样认为的,但是正是这样的认识,差点冤枉了一个研究生。有
一年研究生预答辩时,我看到同组老师的一个研究生ppt中一张蛋白电泳图非常
特别,蛋白条带中间拖后,边缘超前。我就问学生为什么会这样?这个学生说他
也不知道为什么,跑出来的结果就是这样的。maker也是这样。我非常怀疑,但
是保险起见,我没有再争论下去,而是会后自己查了一下,终于发现了原因。他
的蛋白电泳分析的是超低分子量蛋白,用的是Tricine-SDS-PAGE,在这种条件下,
加上电泳仪电压不均衡等其它原因,小分子量蛋白的确会出现边缘超前,中间拖
后的结果。请看网上的一些图片:
http://www.chem17.com/offer_sale/detail/11291438.html
http://www.real-times.com.cn/pic/130912402268636323.jpg
http://img.bimg.126.net/photo/Uy9-b3L0lqS3AQpCra1C8Q==/1689412810218312912.jpg
http://img.diytrade.com/cdimg/1039467/10995309/0/1257489100.jpg
http://muchong.com/html/201301/5442275.html
以上所述,仅就蛋白电泳而言。至于韩春雨文中近500bp大小的DNA电泳条带
不正常的拖尾现象,我暂时没有见到。
此信目的是让您的质疑更加准确无误,以免被人抓住把柄。
(XYS20160729) |
w******g
发帖数: 301 | 28 楼上把sds Page上的western结果用来说明DNA Page,似乎是苹果比梨。 |
P****R
发帖数: 22479 | 29 他说蛋白胶可以出现这种现象,但是DNA胶以他多年经验,应该看不到这种现象。
【在 w******g 的大作中提到】
: 楼上把sds Page上的western结果用来说明DNA Page,似乎是苹果比梨。
|
P****R
发帖数: 22479 | |
|
|
P****R
发帖数: 22479 | |
q*d
发帖数: 22178 | 32 卧槽,每个给方舟子写信的,
开头都是一堆肉麻,结尾又是一堆马屁,
看多了有想吐的感觉
【在 P****R 的大作中提到】
: 关于电泳条带的拖尾问题
: 方先生:
: 您好!
: 我是您的超级粉丝,从“基因皇后”事件就开始关注新语丝,一直到现在,
: 只要打开浏览器,肯定要到新语丝上看一看。
: 看了7月20号关于韩春雨的文章,不谈其它,我只就电泳条带的拖尾现象谈
: 一下我的认识。
: 一般情况下,电泳条带拖尾状况正如方先生所说,边缘拖后,中间超前。我
: 原先也是一直是这样认为的,但是正是这样的认识,差点冤枉了一个研究生。有
: 一年研究生预答辩时,我看到同组老师的一个研究生ppt中一张蛋白电泳图非常
|
{kind=link}
{kind=link}
{kind=link}